Drag a drug into a bin. On phones: tap the drug, then tap its bin.
Classify the Beta-Blockers
Cardioselective (B1 only) vs Non-selective (B1+B2) vs Mixed (B+alpha)
Metoprolol
Atenolol
Bisoprolol
Esmolol
Nebivolol
Propranolol
Nadolol
Timolol
Sotalol
Carvedilol
Labetalol
B1 Only
B1 + B2
B + Alpha
Why B1 selectivity matters
Cardioselective drugs spare B2 at normal doses. B2 lives in bronchioles and peripheral vessels. Blocking it causes bronchospasm and masks hypoglycemia recovery. Cardioselective agents are safer in COPD, PVD, and diabetics. Selectivity is relative: at high doses, cardioselective agents start blocking B2 too.
Decision Tree
Patient needs a beta-blocker. Walk the chain.
A patient walks into clinic needing a beta-blocker. Your job: pick the one that does not kill them. Each branch teaches a different rule.
Step 1. Does the patient have asthma or COPD?
Yes · reactive airway disease
No · lungs clear
Non-selective beta-blockers are contraindicated. Blocking B2 in bronchioles strips the bronchodilator tone and triggers bronchospasm. If a beta-blocker is unavoidable (post-MI, severe HF), use a cardioselective agent (metoprolol or atenolol) at the lowest effective dose with extreme caution. Esmolol IV for short-term in the ICU. Avoid propranolol, nadolol, timolol, carvedilol, labetalol.
Lungs clear, B2 blockade is not a deal-breaker. Move to indication-based selection.
Rule. The B2 receptor on bronchiole smooth muscle is what keeps the airway open. Blocking it during bronchospastic disease can be fatal. Cardioselectivity (B1 over B2) is dose-dependent. At high doses, even metoprolol starts to hit B2. So: low-dose, cardioselective only.
Step 2. Does the patient have systolic heart failure (HFrEF)?
Yes · EF below 40%
No · EF preserved
Use carvedilol (alpha + beta block, also antioxidant) or metoprolol succinate (extended-release, B1-selective) or bisoprolol. Do NOT use propranolol in HFrEF: too much negative inotropy, no anti-remodeling data. Start LOW, titrate slow, only when patient is euvolemic and stable. Acute decompensation is a contraindication to starting.
No HFrEF, no remodeling concern. Move to next indication.
The HF trio: carvedilol, metoprolol succinate, bisoprolol. These are the only beta-blockers with mortality benefit in HFrEF (Class I, level A). Metoprolol tartrate (immediate-release) does NOT have the data; only succinate (XR) does.
Step 3. Recent MI (within 1 year)?
Yes · post-MI
No · primary prevention
Metoprolol succinate is first-line post-MI for mortality benefit. Carvedilol also acceptable, especially with concurrent HFrEF. Reduces reinfarction, sudden cardiac death, and remodeling. Start within 24 hours if hemodynamically stable.
No recent MI. The choice now depends on the dominant indication: HTN, arrhythmia, migraine prevention, essential tremor, performance anxiety, portal HTN, or thyroid storm. Pick by mechanism, not by reflex.
Final rule. Beta-blocker selection is never generic. Match the drug to the indication: HFrEF gets the trio, post-MI gets metoprolol succinate, thyroid storm gets propranolol (blocks T4 to T3 conversion), HTN emergency in pregnancy gets labetalol, portal HTN gets nadolol or propranolol, performance anxiety and essential tremor get propranolol, glaucoma gets timolol topically. The drug name is downstream of the diagnosis.
The Trap
clinical medicine love giving you a patient with COPD AND HF AND HTN. The asthma rule beats the HF rule. If you cannot tolerate ANY B2 block (severe asthma), no beta-blocker is the right answer, and you swap to an alternate (ARB, dihydropyridine CCB for HTN, ivabradine if you only need rate control in HF). Selectivity is your friend, but not your savior.
Glucagon Bypass
Why atropine failed. Why glucagon saved him.
Propranolol saturates the B1 receptor. NE cannot bind. The downstream Gs-cAMP-PKA chain goes silent. Heart rate and contractility fall.
HF Paradox
Beta-blockers cause acute decompensation if started during active HF, yet long-term use reduces mortality. Chronic sympathetic activation causes B1 downregulation and myocyte apoptosis. Beta-blockers interrupt this. The anti-remodeling benefit only appears after 3 months of titrated dosing. Carvedilol, metoprolol succinate, and bisoprolol are Class I in stable HFrEF. Starting during decompensation is contraindicated for the same reason they work long-term: they acutely reduce contractility.
What It Looks Like
EKG, receptor, molecule. Tap to expand.
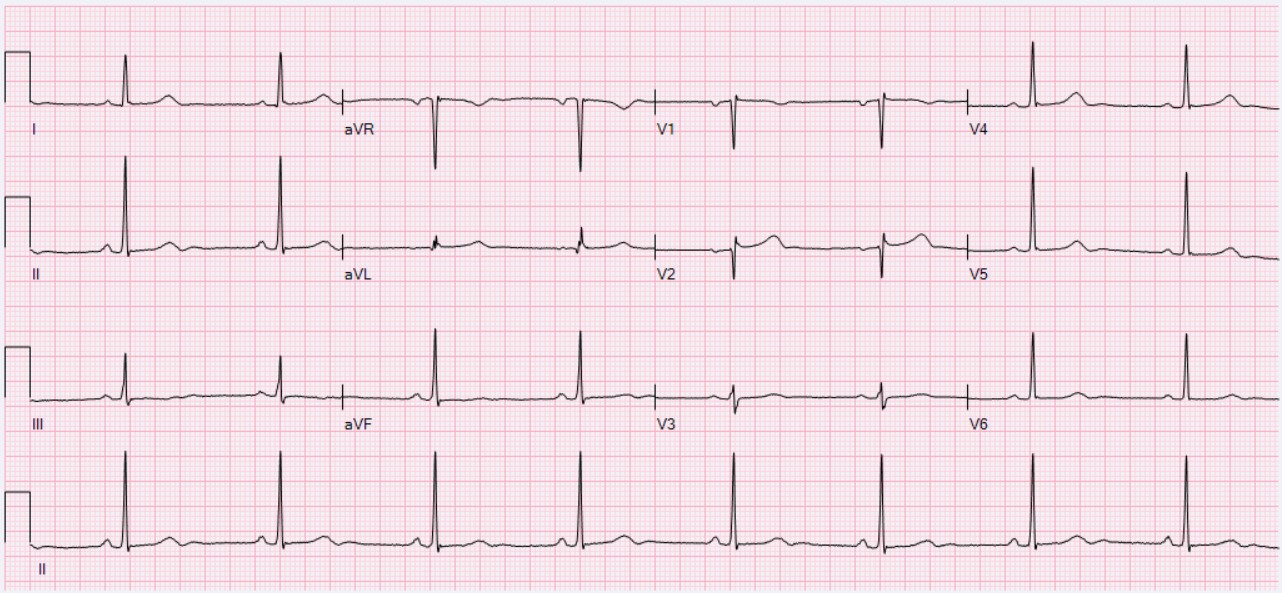
EKG · sinus bradycardia
Three anchors. The EKG is what your patient looks like on a beta-blocker (rate down, PR sometimes up, no other changes unless toxic). The receptor is the seven-transmembrane GPCR that propranolol fills, denying NE its binding site. The catechol ring plus ethylamine tail is what every endogenous agonist (NE, EPI, DA) shares; beta-blockers mimic enough of this scaffold to compete for the orthosteric site.
Test It
10 questions. Read every explanation.
32-year-old male, propranolol OD. HR 38. IV atropine x2: no effect. Why did atropine fail?
Tempting to give more atropine: bradycardia on an ECG looks cholinergic, and atropine is the reflex drug for slow heart rate. The trap is that propranolol-induced bradycardia is adrenergic, not cholinergic. Atropine blocks muscarinic (ACh) receptors, which are not involved here at all. Think of two separate brake pedal systems in a car: atropine releases the cholinergic brake, but propranolol has locked the adrenergic brake from the other side. Releasing one brake does nothing when the other is padlocked. Bradycardia is adrenergic, not cholinergic, which is why atropine fails. Fix: glucagon (bypasses B-receptors via cAMP) or high-dose insulin euglycemic therapy. Break it down: propranolol OD bradycardia = adrenergic mechanism (B1 block); atropine = muscarinic/cholinergic block only, useless here; treatment = glucagon or high-dose insulin, not more atropine.
Patient with mild COPD and newly diagnosed hypertension needs a beta-blocker. Best choice?
Tempting to avoid beta-blockers in COPD entirely, or to pick propranolol as the most familiar agent. Propranolol and nadolol are non-selective: they block B2 in the bronchi and can trigger bronchospasm. Think of cardioselective beta-blockers as precision screwdrivers that only fit the B1 cardiac screw, leaving the B2 bronchodilator screws alone. Metoprolol succinate (B1-selective) can be used in mild-moderate COPD with appropriate monitoring. Sotalol is non-selective and adds QT prolongation risk with no benefit here. Break it down: COPD + beta-blocker = cardioselective only (metoprolol, atenolol, bisoprolol); propranolol, nadolol, sotalol = non-selective = B2 blockade in lungs = bronchospasm risk; cardioselectivity is relative, not absolute.
Patient in thyroid storm. Propranolol is chosen over other beta-blockers. Which TWO mechanisms justify this?
Tempting to include "reduces T3/T4 synthesis" as a propranolol mechanism since it is given in thyroid storm. That is the mechanism of PTU and methimazole. Propranolol cannot touch thyroid hormone synthesis. Think of propranolol in thyroid storm as a fire brigade that cannot extinguish the furnace but can stop the fire from spreading: it controls the adrenergic symptoms via non-selective beta blockade AND blocks the peripheral conversion of T4 to the more potent T3 (deiodinase inhibition). Non-selective B-blockade is required for the T4-to-T3 blockade, which is why cardioselective agents are not adequate here. Break it down: propranolol in thyroid storm = (1) non-selective B-block controls adrenergic symptoms; (2) inhibits peripheral T4-to-T3 conversion; does NOT reduce synthesis; cardioselective agents lack the T4-to-T3 mechanism.
Stable HFrEF (EF 30%), started on carvedilol. At 2 weeks EF is 28% and symptoms are slightly worse. Correct interpretation?
Tempting to stop carvedilol: EF dropped, symptoms worsened, that looks like drug harm. The trap is that early worsening is expected and stopping the drug is the wrong call. Think of beta-blockers in heart failure like physical therapy after knee surgery: the first two weeks feel worse than before surgery, but stopping PT at week two throws away the long-term benefit. The negative inotropy of carvedilol is a short-term cost that produces anti-remodeling benefit over months: reverse remodeling, improved EF, and reduced mortality. Stable hemodynamics are the only prerequisite to continuing. Break it down: beta-blockers in HFrEF = expected early worsening (negative inotropy); anti-remodeling benefit appears at 3 to 6 months; never stop for early symptom worsening if hemodynamically stable; the three approved agents are carvedilol, metoprolol succinate, and bisoprolol.
Type 1 diabetic on insulin has a hypoglycemic episode while on propranolol. Which symptom is most likely MASKED?
Tempting to pick diaphoresis or tremor as the masked symptom since beta-blockers seem to blunt all catecholamine effects. The key distinction: sweating in hypoglycemia is mediated by acetylcholine (cholinergic), not by the adrenergic pathway that beta-blockers block. Think of beta-blockers as silencing one specific alarm in a house: the heart-rate alarm (adrenergic) is cut, but the sweating alarm (cholinergic) still rings because it uses a completely different circuit. Tachycardia is the warning sign that gets silenced, allowing the glucose to fall further without the patient noticing. Break it down: propranolol masks tachycardia (adrenergic) in hypoglycemia; sweating is preserved (cholinergic/ACh, not affected by B-block); hunger and confusion are also preserved; the masked tachycardia is what makes beta-blockers dangerous in insulin-dependent diabetics.
A 48-year-old with stable angina and well-controlled HTN is started on a beta-blocker. He also has Raynaud-like episodes after starting the drug, but his exercise tolerance has improved. Which agent has intrinsic sympathomimetic activity that could have prevented the cold extremities and could be considered if symptoms persist?
Tempting to pick a cardioselective agent (metoprolol, atenolol) on the theory that sparing B2 in the periphery reduces vasoconstriction. That logic is partially right but misses the specific question: which agent has intrinsic sympathomimetic activity (ISA). ISA means the drug is a partial agonist at beta receptors, providing a floor of activity rather than complete blockade. Think of pindolol as a thermostat set to minimum heat: even when you turn it "off," it keeps a baseline warmth going in the peripheral vessels via partial B2 agonism. Metoprolol and atenolol have no ISA. Propranolol is non-selective with no ISA, worsening vasoconstriction. Break it down: ISA = partial agonist at beta receptors; pindolol and acebutolol have ISA; ISA prevents resting bradycardia and peripheral vasoconstriction; useful when complete beta blockade causes excessive cold extremities or bradycardia.
A 58-year-old comes in with chest pain at rest, ST elevations that resolve after IV nitroglycerin, and a clean coronary angiogram. He has been taking propranolol for migraine prophylaxis. Why might propranolol have made this worse?
Tempting to think propranolol would help coronary chest pain since it reduces oxygen demand and heart rate. The trap is that Prinzmetal angina is vasospasm-driven, not demand-driven. Think of coronary artery tone as a tug-of-war: B2 agonism pulls toward vasodilation, alpha-1 pulls toward vasoconstriction. When propranolol blocks B2, it removes the vasodilation team entirely while leaving the alpha-1 vasoconstriction team at full strength pulling alone. This leaves alpha-1-driven coronary vasospasm unopposed. Propranolol causing headaches (migraines) and then coronary spasm is a classic clinical medicine double-trap. Break it down: beta-blockers are CONTRAINDICATED in Prinzmetal/vasospastic angina; B2 blockade removes vasodilatory counterforce and leaves alpha-1 vasoconstriction unopposed; calcium channel blockers (not beta-blockers) are the treatment for vasospastic angina.
A 45-year-old with cirrhosis has esophageal varices on EGD. The hepatologist starts a non-selective beta-blocker for primary prophylaxis against variceal hemorrhage. Which mechanism explains the benefit?
Tempting to pick B1 blockade alone as the mechanism: lowering HR and CO seems like enough to reduce portal pressure. The trap is that the splanchnic vasoconstriction component requires B2 blockade too. Think of portal hypertension inflow as two pipes: the cardiac output pipe (B1 block reduces flow from the heart) and the gut circulation pipe (B2 block lets alpha-1 constrict the splanchnic vessels, reducing inflow from the bowel). Cardioselective agents only close one of the two pipes. Non-selective agents (propranolol, nadolol, carvedilol) close both. Direct hepatic venous dilation is not the mechanism. Break it down: non-selective BB for varices = B1 block lowers cardiac output plus B2 block allows alpha-mediated splanchnic vasoconstriction; both mechanisms together reduce portal pressure; cardioselective agents are NOT effective for variceal prophylaxis.
A 28-year-old G2P1 at 34 weeks gestation arrives in triage with BP 195/120, headache, and proteinuria. The team needs to lower her BP urgently without harming the fetus. Which beta-blocker is first-line for hypertensive emergency in pregnancy and why?
Tempting to pick esmolol: ultra-short half-life sounds safe since it wears off quickly. The trap is that esmolol crosses the placenta and causes fetal bradycardia and respiratory depression. Think of placental crossing like a shared water supply: esmolol gets into the fetal circulation fast and causes the same B-blockade in the fetus that you want in the mother. Labetalol is the first-line choice because it combines alpha-1 afterload reduction with beta blockade, lowering BP without reflex tachycardia, and has an established fetal safety profile at therapeutic doses. Atenolol is associated with fetal growth restriction when used chronically. Break it down: labetalol = alpha-1 plus B1 plus B2 blockade, afterload reduction without reflex tachycardia, first-line for hypertensive emergency in pregnancy; esmolol = fetal bradycardia and respiratory depression risk; atenolol = fetal growth restriction; metoprolol used in pregnancy but not first-line for acute emergency.
A 24-year-old violinist auditions for a major orchestra and develops palpitations, tremor, and dry mouth before each audition. He has no asthma, no HF, no DM. Which drug provides symptom control without sedation, and what receptor effect drives the tremor reduction?
Tempting to pick atenolol since it is cardioselective and causes less central sedation than propranolol. The trap is that physiologic tremor is driven by B2 receptor activation in skeletal muscle spindles, and atenolol is B1-selective and does not reach the B2 receptors in muscle. Think of skeletal muscle tremor as a B2-powered vibration motor: only a drug that can reach and block that specific B2 motor will quiet it. Propranolol penetrates muscle B2 receptors as a non-selective agent. Lorazepam would cause sedation, which is exactly what a performing musician cannot have. Break it down: propranolol for performance anxiety = B2 blockade in skeletal muscle spindles reduces physiologic tremor amplitude; atenolol = B1-selective = misses the B2 tremor mechanism; lorazepam = sedation = wrong for performers; propranolol does not impair cognition at low doses.
clinical Walkthrough
clinical Walkthrough
Original clinical vignettes. Shuffled, never-repeat, full explanations for every choice.
Medically reviewed by Kaitlyn Cocuzzo, MD and Fatima Ali, DO · Last updated July 5, 2026 at 8:04 PM ET
Bone Wizardry is an independent educational resource for visual learning in the medical sciences. It is not affiliated with, endorsed by, or sponsored by any licensing or examination board, contains no real or recalled examination questions, and does not guarantee any educational or examination outcome.